离聚物(离子交联聚合物)是一种聚合物,即一长串的重复单元,其他类似结构的例子如蛋白质和橡胶等,用来作为AEM膜本身和催化剂(CL)的粘合剂,也即配制浆料,形成MEA。离子聚合物由通过共价键连接到主链上的离子化和电中性单元组成。AEM水电解槽和燃料电池需要离子聚合物将OH-电荷载体从膜上传输到催化剂的活性位点(取决于阴极/阳极是否被考虑),同时通过机械方式将催化剂层粘合在一起,同时在CL中形成孔隙网络。离聚物还有助于将整个CL固定在底层基底上,从而提供机械稳定性并延长ECSA的使用寿命。离子聚合物的理想含量是一个需要优化的问题,因为离聚物是离子载体,其电子导电性不足。离子聚合物含量过多会阻碍电子传输,含量过少,氢氧根离子可能无法到达活性催化位点。 水电解制氢设备
离聚物的选择通常取决于 AEM 的类型,因为使用与AEM相似的离子载体并避免竞争性化学环境整体是有利的 。但是,由于 AEM 和离聚物的作用不同,不能使用液态AEM 作为离子交换膜。具体来说,AEM需要具有较低的气体渗透性,以抑制氧气和氢气的渗透,而离聚物则需要具有较高的气体渗透性,以便迅速排除产品气体,从而避免活性催化剂位点的堵塞。
目前市面上已有几种商用离聚物,主要包括杜邦公司的Nafion®、索尔维公司的Aquivion、迪奥克斯公司的Sustainion、艾奥姆公司的Aemion、维索根公司的PiperIon、杜瑞安[和Xergy公司的Pention、富马泰克公司的Fumion以及东丽公司的Tokuyama。除了商业产品外,实验用离聚物也经常被报道。
Tokoyama 是十多年前最早提供商用 AEM 和离聚物的公司之一 ,由于该品牌已停产,导致最近很少使用他们的产品。当阳极制备为CCM时,当催化剂从膜上分离脱落时,会产生不良反应,这种影响归因于AS-4离聚物(Tokuyama)的机械/化学稳定性差。然而,在与水合作用相关的溶胀效果方面,Tokuyama 优于 FAA-3 Fumion 离聚物 (FuMA-Tech) 。
除了与AEM的化学成分相匹配外,由于吸水率(WU)的增加,选择具有高离子交换容量(IEC)的离聚物已被证明有利于在使用干阴极运行(差压式结构)时保持阴极湿度。IEC和WU的方面也是很复杂的,因为通常伴随着显著的IEC/WU比率,这会导致过度溶胀,这可能会通过移除催化剂颗粒来破坏CL的机械完整性,并降低ECSA并增加ICR。
考虑到阳极/阴极的不同电化学环境,以及水在其中作为产物/反应物的相反作用,离聚物的作用和影响将根据考虑的电极而变化。水作为产物在阳极上的作用非常重视疏水性,以避免 MT(质量传输) 问题。为此,早期的离聚物优化研究利用阳极 CL 中少量添加的电中性 PTFE 来充分解决这些问题。将理想化的含量被量化为 9wt.%,从而能够形成必要的孔隙结构,但不会遭受到 PTFE 的绝缘效应从而增加欧姆电阻。随后的研究以及验证了110wt.% 的范围,并证实了这一数量的合理性。 高纯氢气发生器
在NiO催化剂(5mgNiO/cm2)的条件下,对Fumion FAA-3离子聚合物的使用量进行了系统的调整,在10~40wt.%的范围内变化,但未发现对性能和团聚程度有明显影响。然而,后来出现了相反的结果,在与镍纳米粒子(5mgNi/cm2)一起研究7~25wt.%的范围内时,15wt.%时的相同Fumion离聚物的OER活性达到峰值。该载量足以提供低的电荷转移电阻和良好的附着力,同时低到足以避免溶胀问题,溶胀会对CL的机械稳定性带来挑战。随后,还对相同的Fumion离子聚合物进行了5、10、20、30和40wt.%的负载量下采用IrO2催化剂(2.0mgIr/cm2)测试,显示10wt.%的负载量下出现明显的峰值。该载荷量下产生了通过LSV测量的最大电流,并辅以低电荷转移电阻和优化的形貌而得到增强。进一步增加离子聚合物含量会增加Rct,并导致MT问题,因为过量的量会堵塞活性位点并减少孔隙体积。对FAA-3离子聚合物的阴极/阳极同时进行调整,在使用IrO2阳极(2.0 mgIr/cm2)和Pt/C阴极(0.40mgIr/cm2)时,在20wt.%时获得了最高电流。
后来,该离聚物与Aemion AP1-HNN8进行了比较,使用7wt.%离聚物和铱黑催化剂(3.5-3.8 mgIr/cm2)。Aemion AP1-HNN8未能达到 Fumion FAA-3 离聚物的水平,因为它未能建立足够紧密的 CL-膜界面,这导致三相边界点减少和阳极的 Ir 物理损失(从75at.%到13~18 at.%)。这个缺点导致催化剂层中的离子电阻增加,从而导致欧姆电阻增加 了1.7倍。对于IrOx阳极CL (1mgIr/cm2),Aemion AP1-HNN8含量在4、7、10和15wt.%之间系统的变化调整,其中在4wt.% 达到峰值活性。然而,相关的稳定性低于7wt.% 的阳极,后者产生了最高的组合性能。其他研究人员重申了这一点,因为离聚物含量必须足够高才能提供结合力和稳定性,但又要足够低以避免由于溶胀和层异质性而导致的动力学/欧姆/MT损失。
将 Aemion AP1HNN8 与 Fumion FAA-3 和 Nafion® 进行比较时,也发现了类似的趋势。使用 NiFe作为 OER 催化剂评估这三种离聚物的性能表明,相对于 Nafion-NiFe 组合(5mgNiFe/cm2),碱性离聚物的性能有所降低。理论上,其原因在于苯基吸附,苯基吸附可以阻断OER 活性位点和/或形成酸性苯酚,从而降低局部 pH 值并降低 pH 依赖的电化学活性。此外,NiFe 催化剂中从 Ni(II)到 Ni(III)的转变有利于动力学改善而提高反应活性,尽管当与Aemion AP1-HNN8 结合时,由于这些吸附效应而使用时受到抑制。
苯基吸附在OER 催化剂上是通过苯基环的 C-H 键裂解而发生的。苯基的氧化程度随催化剂的类型而有很大差异,因为最近的一项研究发现,由于表面相互作用较弱,与La0.85Sr0.15CoO3钙钛矿相比,BTMAOH苯基被Pt/C和IrO2氧化的程度更大。此外,CL 中的大量氧与与离聚物的不良相互作用增强有关。DFT 计算支持苯基氧化与苯基催化剂吸附能之间的相关性,这表明为 AEMWE 应用选择非PGM 催化剂材料具有额外的优势,并阐明了某些 PGM 催化剂材料的长期碱性稳定性差的原因。
几种实验离聚物也显示出潜力,其中将季铵化聚咔唑基聚合物(聚(9-(6-(三甲基溴化铵)己基)-9H-咔唑-1,1,1-三氟异丙烷,简称为QPCTMA)与Fumion FAA-3离聚物进行了比较。与其他发表的研究类似,基于 5~30wt.% 之间的负载量,10wt.% 的离聚物负载量被认为是最理想的,加载增量以5wt.%进行调整。根据固有水需求调整阳极/阴极离聚物的比例是燃料电池的既定做法,但最近才将相同的方法用于电解槽。用IrO2阳极(2.0mgIr/cm2)使用具有高IEC(2.80 mmol/g)的微疏水性(≈80%吸水率)离聚物与干阴极(差压式)结合使用,当使用10 wt.%的离聚物负载量时,对水的整体管理效果显著。
阴极在相关的还原过程中需要水,这意味着需要具有高离子电导率的亲水材料。当CeO2-La2O3/碳载体与镍结合时,将Tokuyama离聚物的载量从5wt.%增加到10wt.%时,过电位下降了47mV(Acta 4030,2.3 mgNi/CeO2-La2O3-C /cm2)。在研究使用Ni/C催化剂的Fumion FAA-3离聚物在10~40wt.%范围内的负载量时,同样确定了10wt.%的载荷为最佳载量。负载量大于10wt.%会增加催化剂的团聚,从而降低了三相界面点的频率、孔体积和活性位点的数量。
一项类似的研究比较了 Pt/C 阴极 (0.40 mgPt/cm2 )与 10、20、30 和 40wt.% 的Fumion FAA-3 离聚物的性能,结果显示30wt.%时出现了明显的峰值活性。直到30wt.%之前电荷转移电阻逐渐降低,但进一步增加导致Rct显著增加,并出现MT过电位。这些效应的出现证实了离聚物作为离子电荷转移介质和 CL 形态创建的作用。
Aemion AP1-HNN8 的阴极离聚物含量为10wt.% 和 20wt.%时,其中前者产生了最佳的性能,尽管改善的幅度与研究人员观察到的对 Fumion FAA-3 离聚物的改善幅度不同。这可能与Aemion离聚物的性能相对较低有关;然而,它也可能受到 CL 制造工艺方法的影响,因为其他研究人员使用 CCM方法,而以上研究采用的是CCS 方法。
在碱性HER反应中,水是反应物,在干阴极(差压式)/无水操作模式下,离聚物的使用最为重要,因为水是碱性HER期间的反应物。将具有高 IEC(3.43mmol/g)的高亲水性(≈350% 吸水率)离聚物与 Pt/C 阴极 (0.50 mgPt/cm2)配合一起使用,它与微疏水性阳极离聚物和具有高水扩散率的 AEM可有效协调。研究了 10 、 20 、 25 、 30 和 40wt.% 的离聚物含量,揭示了 PFBP 离聚物的 25wt.% 载量处出现峰值。这导致在 80°C、2.0V 时产生 7.68A/cm2 的峰值电流密度,以及在1.0M KOH电解液和相对苛刻的去离子水中产生高的AEMWE性能。高水扩散率允许反应物在阳极到阴极之间持续的输送反应物,这为在1.0M KOH 电解质和相当苛刻的去离子水中实现这种高 AEMWE 性能创造了有利条件。
由于具有内在微孔结构(PIM)的聚合物,由于其高电导率 (σ > 150 mS/cm) 和灵活的设计特性,该聚合物已被研究作为为AEMs;然而,理论上微孔结构可能会导致较高气体扩散速率,从而导致气体交叉渗透的可能性很高。因此,PIM可能不适合用于制造AEM,但非常适合做离聚物。用三氟甲基或氰化物取代的苯甲醛聚合聚苯烷烃可产生几种高孔隙率的离聚物,其中一些含有促进孔隙形成的螺吡吲啶结构(spirobisindane structures)。在不同的 AEMWE 电解池中,共有 8 种不同的离聚物用作阳极和阴极离聚物,负载量均为 0.75mg/cm2,其中具有螺吡吲啶结构(spirobisindane structures)离聚物尽管具有相似的 IEC 但产生了最佳性能。通过测量 ECSA,与用氰化物取代的离聚物相比,具有三氟甲基取代离聚物的 AEMWE 显示出更大的表面积。这归因于具有螺吡吲啶结构(spirobisindane structures)的 CF3 取代离聚物提供的更大自由体积,它优化了离子、水和气体在三相界面中的传输。还在1.0M NaOH (j=1.0A/cm2 ,T= 80°C) 中评估了其耐久性,其中具有螺吡吲啶结构(spirobisindane structures)的CF3取代的离聚物 (QP1-CF3-3) 在 180小时稳定性测试中没有表现出明显的长期降解。随后的 LSV 曲线也证实了与稳定性测试之前获得的性能几乎相同。在评估这些稳定性测试的结果时,离聚物是随着时间的推移通常导致失效的成分。这极大地影响了离聚物的稳定性。
阴离子交换离聚体(AEI)的稳定性是 AEM 水电解槽和燃料电池领域最热门的研究课题之一 。离聚物本身在碱性或 pH 值中性环境中的稳定性,或其与各种催化剂在任一电解质中结合的稳定性等课题,似乎都是研究不完的,因为持续有大量的材料被创造出来。
根据对基本机制进行详细研究的趋势分析,目前已经对 AEIs 和铂的结合进行了大量研究,已经证实铂会受到较常见的电荷载体基团季铵(QA)的不利影响。这种影响是通过抑制催化剂活性或铂与 QA 之间的特异性/共价作用产生的。活性位点阻塞的严重程度随着烷基链长度的增加而增加,考虑电荷载体基团的选择顺序如下:四甲基 < 四乙基 < < 苄基三甲基 。苄基三甲基铵的苄基与电极表面之间的强烈相互作用导致其对活性位点阻塞的影响最为显著。这种相互作用有两种可能的吸附过程:
(1) 阳离子-羟基-水共同吸附在活性位点上,或
(2) 苯基的吸附阻止了进一步的催化活性。
由于离聚物的作用是提高界面之间的离子电导率,因此考虑双层模型可能会有所启发,在研究 AEI 与各种铂表面之间的相互作用时就采用了这种方法。为此,对 Pt-Nafion 和 Pt-AEI 进行了循环伏安法分析,其中初始电位范围从 0.05~0.50 VRHE 逐渐扩展到 -0.10~0.50 VRHE。初始范围在阳极扫描上产生源自氢解吸(欠电位沉积氢 Hupd)的常规峰值,两种材料系统之间没有差异。当电位范围扩展到 HER 电位时,两个系统之间出现了明显差异,因为观察到与过电位沉积氢 (Hopd) 相关的峰值存在巨大差异。理论上,Hopd本身的来源于氢氧化产生的法拉第电流和 Hupd 伪电容电荷的叠加,或者来自 HER 中间体的其他单层的伪电容。无论来源如何,两个系统之间 Hopd 电荷的差异并不意味着AEI起始部分的特异性吸附性导致 ECSA 降低,而是由于它们的接近而降低了HER性能。
这表明 AEI 同时影响内亥姆霍兹平面和外亥姆霍兹平面(IHP 和 OHP),其中大多数 AEI 部分都在OHP 中。IHP 和 OHP 中 AEI 的存在也与早期关于甲醇氧化反应 (MOR) 的研究相关,其中氧化电极的电位自然为正(相对于零形式电荷 (PZFC) 的电位),这意味着带正电荷的 QA 阳离子将被排斥。在存在 AEI 部分的情况下,MOR 性能降低与 OHP 的较高有效电位 (Φ2) 有关,因为 AEI 的化学电位引起的静电效应会导致Φ2增加 ,如下图1(a) 所示。这限制了 OH– 向 IHP 的重要运输,而 IHP 是去除 MOR 副产物 COads 所必须的。同样,对于 HER来说,AEI 将增加 Φ2 (图1(b)),这意味着需要更大的电极电位 (ΦM) 才能在电极表面之外感应出相同的电位。因为电极溶剂化壳也是反应物,这相当直接地作用于 HER,这意味着 IHP 电位 Φ1 将决定速率常数。
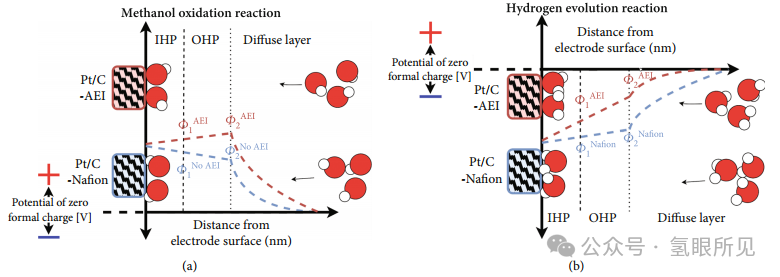
一般来说,对于离聚物在实际操作条件(E≤2.0V, j≥1.0A/cm2 , T≥50°C)下的相关稳定性的研究很少,因为它们在相当温和的条件下的稳定性仍在研究中。有研究报道了Fumion FAA-3和Durion Orion TM1离聚物在0.5A/cm2、50~80°C温度下连续运行时的稳定性。尽管 Orion TM1 离聚物的性能优于Fumion FAA-3,但报告仍显示了其明显的电压衰减率,特别是在温度为 50、60、70 和 80℃时,衰解率分别为 40、47、55 和 321mV/h。使用 FAA-3 离聚物和 AEM 的配合在 3.4 小时内电压迅速增加到 2.3V,从而不得不结束测试。其他研究也报告了类似的衰减率,研究中 FAA-3 离聚物与 IrO2/NiFe 阳极以及 Pt/C 阴极一起使用。AEMWE单电解池在工作温度70°C 下,基于CCM工艺的IrO2 和 NiFe 阳极以1.0A/cm2运行的情况下性能分别下降了 22.7和16.7mV/h,在基于CCS 的工艺中分别降低了22.0和11.0 mV/h。
使用1.0M K2CO3/KHCO3缓冲液(pH10)、1.0M KOH(pH14)或1.0M硼酸盐缓冲液(pH8)作为电解质溶液,评估了市售商用离聚物Aemion AP1-HNN8-00-X(离聚物)、Sustainion-XA9(二氧化物材料)和PiperIon PAP-TP-85(Versogen)组成的薄膜的稳定性。为了简化分析,薄膜不含催化剂材料。
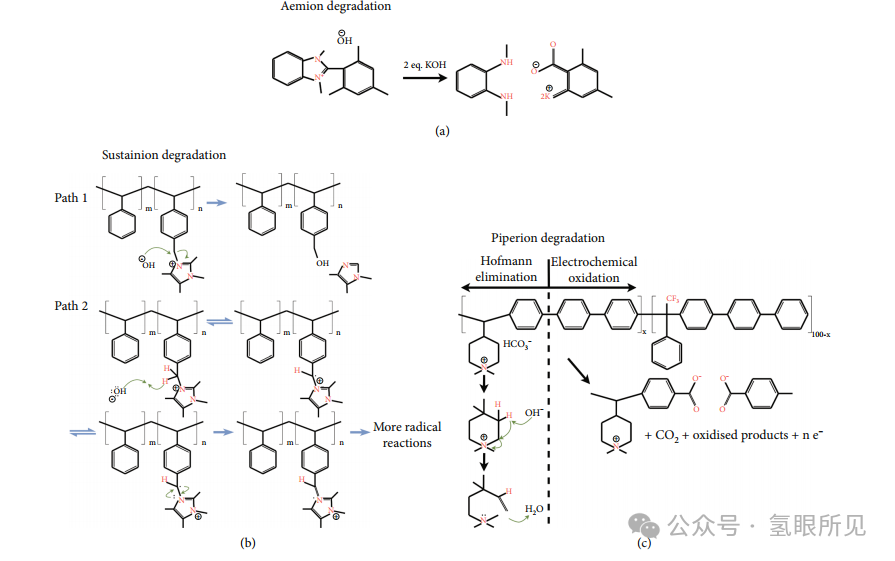
Aemion AP1-HNN8-00-X 在所有电解质中都会发生结构重组,但在 K2CO3/KHCO3 缓冲溶液中会完全溶解。基于石英晶体微天平测试期间离聚物的完全损失以及在高结合能C 1s 峰的出现 ,推测聚合物主链通过逐渐断裂成可溶性碎片而降解。这可能是由于电荷载流子基团的位置所致,因为它位于骨架主干本身,而不是连接到主干的侧链上。 PEM制氢设备
如下图2(a) 所示,预计 Aemion 容易受到亲核攻击,其中苯并咪唑带电基团的开环和源自聚合物的羧酸盐的形成导致离聚物薄膜的溶解。这可能是由于带电的邻近电极降低了电子密度,导致电解质中的 OH– 电荷载流子变得具有强亲核性,进而使得Aemion 离聚物降解,因为它通过明显的甲基化得到了稳定。衰减全反射 (ATR) FTIR 光谱分析证实了这一点,其中 753/cm附近出现的与芳香族化合物变化相关的新模式表明,由于亲核攻击,电荷载流基团中的氮原子发生了去甲基化。此外,ATR-FTIR 还揭示了 C=N 模式强度的降低,显示了N+ 的丢失,XPS 结果也表明了这一点。
Sustainion在 K2CO3/ KHCO3 缓冲液中,显示出更高的稳定性,理论上这源于两种离子聚合物的碳酸盐/碳酸氢盐离子传导性存在固有差异。离聚物能够快速传导阴离子,从而避免在离聚物/电极界面中形成 pH 值或浓度梯度。然而,虽然离聚物主链保持完整,但电极表面上残留的薄膜表明电荷携带基团的稳定性也受到了挑战。
如下图2(b)所示,咪唑鎓(imidazolium)电荷载流子基团在亲核攻击下开环,XPS 测定的总 C-N/C 比值的降低表明以咪唑鎓(imidazolium)基团为末端的侧链消失了。ATR-FTIR 光谱也证明了这一点,该光谱显示了 C=N 模式的变化。根据 N 1s光谱显示的咪唑( imidazolium)基团氧化态的变化,侧链的脱落很可能发生在咪唑基( imidazolium)团重组之前。环的打开可能是侧链丢失的过渡阶段,或者如下图2(b)中的路径 1 和路径 2 可能通过亲核攻击同时发生。重要的一点是,在没有施加电位的情况下,Sustainion 离子聚合物不会明显降解,这表明亲核攻击必须通过氧化电位来加剧。此外,OER 中间体(如、HO2 和 HO)可能会氧化连接咪唑( imidazolium)和离子聚合物骨架的苯基,从而导致导电性降低。
与 Aemion 类似,PiperIon 也容易受到 K2CO3/KHCO3 缓冲液的影响。/
阴离子的低电导率可能会引起离聚物的电化学氧化,也可能会受到薄膜/电解质界面的浓度或 pH 梯度造成的局部 pH 值降低的不利影响。电化学氧化可能会影响连接电荷载体和聚合物骨架的苯基;不过,这种降解很难得到证实,因为降解产物可能已经溶解。在纯水 AEMWE 中,PiperIon 与 IrO2 一起工作时会发生大量氧化。在这里,XPS 测定的 C-N 键的数量大大减少,这一点通过不存在的 N 1s 光谱得到了证实。此外,F 1s光谱被削弱,表明三氟甲基的消失。采用密度泛函理论 (DFT) 计算以确定三种离聚物的氧化还原电位如下:( 2.18 eVSHE cis, 2.33 eVSHE trans > PiperIon 1.78eVSHE > Aemion 1.46eVSHE )。Sustainion受到与π堆叠相关的竞争效应的影响,特别是基态失稳和通过多中心单电子键稳定自旋密度。
考虑到氧化还原电位较低,因此假定 Aemion 会被氧化。具体来说,苯并咪唑(benzimidazolium )中没有附着甲基的氮原子是最脆弱的点,因为甲基增加了自旋稳定性。因此,如下图2(c)所示,电化学极化辅助苯并咪唑()电荷载体基团开环被认为是 Aemion AP1-HNN8-00-X 离聚物的主要降解途径。这可能是通过对极化离聚物结构的亲核(OH-)攻击,也可能是由于 OER 中间体和/或电极表面的直接氧化导致。对于 Sustainion 来说,电荷载体提供的保护会稳定邻近的苯基环( phenyl ring),从而使另一个临近的不含咪唑鎓(imidazolium)的苯基环更容易成为氧化目标。然而,氧化还原电位很高,而且实验证据表明氮键环境发生了变化,这在苯基氧化中是不存在的。有鉴于此,连接苯基和电荷载体基团的 C-C 键被氧化,导致侧链脱落,最有可能是降解的方式。
当催化剂和离聚物结合时,活性和稳定性会因系统而异,最终产生无穷无尽的组合。因此,发现可靠的趋势将对减少为 AEM 电解槽应用制造稳定离子膜所需的工作量产生重大影响。最近再次研究了市售离聚物 PiperIon PAP-TP-85 的稳定性,这次与一系列过渡金属氧化物一起进行了研究,并针对 IrO2 进行了研究 。发现具有高电子导电性的催化剂会产生更高的降解速率。Co3O4 和 IrO2 作为导电性最强的材料,在去离子水中20小时的降解率分别保持在1.8和2.6mV/h (j=1.0A/cm2 ,T=57°C)。然而,催化剂最大的降解原因与催化剂重构有关,其中反复溶解和再沉积改变了离聚物-催化剂之间的相互作用并阻断了活性位点。由于催化剂离聚物相互作用存在的问题,一些研究选择不使用离聚物,而是直接在 GDL 本身中产生CL(比如真正意义的原位生长或者类似现状传统碱性电极的做法)。在AEMWE单电解池中测试了几种不含离聚物的MEA,在1.0M KOH中放置120小时后,峰值稳定性为0.91mV/h(j=0.4A/cm2,T=40°C)。虽然降解率相当低,但工作条件和性能仍低于与成熟的 PEM 和传统 AWE 技术竞争所必需的性能条件。此外,工作电压 (≈2.20 V) 略高于其他选择使用离聚物的装置所显示的电压,这表明在 CL 中包含此类离子导电元件可以进一步改进。
有关聚降冰片烯(polynorbornenes )在 AEMFC 中的稳定性和离子导电性能的报道也屡见不鲜,但最近才有关于 AEMWE 离聚物的报道。最近报道了一种新的实验性离聚物,它将催化剂、AEM、GDL 和非离聚物环氧树脂粘合剂共价键合在一起,对提高稳定性有很好的效果。虽然最初通过增加非离聚物粘合剂的用量提高了稳定性,但只是因为粘合剂的化学键将催化剂和离聚物固定在了一起,稳定性才得到增强。此外,当催化剂包裹在粘合剂中时,离聚物无法与催化剂和 AEM 粘合,从而降低了 MEA的离子传导性并降低了其稳定性。新型 TP2 离聚物由长度可观的取代基链(-CH2-CH2-COOH 取代基)组成,这使得离聚体与环氧粘合剂之间形成共价键具有更大的灵活性,从而提高 CL 中的离子电导率。AEMWE 在 1.0 A/cm2 、60°C 的 0.1M NaOH 中运行了500 小时,证明了离聚物的稳定性,也没有观察到的降解。通过比较稳态 LSV 曲线观察到了性能的提高,这是与 NiFe2O4 阳极催化剂的磨合期延长有关。
与具有芳基-芳基主链的市售聚芳基(poly(aryl(piperidinium))/(PAP))离聚物相比,使用具有烷基-烷基(aryl-aryl)链的聚降冰烯(polynorbornene)离聚物的实验离聚物显示出更高的稳定性。据报告,使用此类离聚物的优化 AEMWE 的总体降解率为 8.3 μV/h。不同的哌啶( piperidinium)离聚物(poly(fluorenyl-co-aryl piperidinium) (PFAP))在 1000 小时(j=0.5A/cm2 ,T=80°C)内表现出良好的稳定性,其中1H 核磁共振显示出完整的骨架和电荷载流基团。同样,三氟甲基取代的苯甲醛聚合聚(phenyl-alkane)离聚物在1.0M NaOH 中180小时后没有表现出明显降解(j=1.0A/cm2,T=80°C)。另一方面,氰化物取代苯甲醛聚合的聚 (phenyl-alkane) 离聚物在120小时后出现不可恢复的损失,在剩余的 60 小时稳定性测试中,电解池电压突然从1.75V 升至 1.85V 。

